Cognitive Assessment and Neuroimaging of Dementia
Bob Chaudhuri, MD
Department of Psychiatry,
University of Toronto,
Toronto, ON.
Contributions from:
Morris Freedman, MD, FRCPC
Director, Behavioural Neurology
and Senior Scientist,
Rotman Research Institute,
Baycrest Centre for Geriatric Care,
Professor of Medicine (Neurology),
University of Toronto,
Toronto, ON.
Larry Leach, PhD, CPsych
Psychologist, Department of Psychology,
Baycrest Centre for Geriatric Care,
Adjunct Professor,
University of Toronto,
Toronto, ON.
Wendy Meschino, MD, CCFP, FRCPC, FCCMG
Clinical Geneticist,
North York General Hospital,
Toronto, ON.
On Sunday, March 18th, a series of speakers discussed cognitive assessment and neuroimaging in dementia. The speakers included Dr. Morris Freedman, Dr. Larry Leach, Dr. Robert van Reekum, Dr. Sandra E. Black and Dr. Wendy Meschino.
The focus of Dr. Freedman's workshop was the Bedside Assessment of Cognitive Function. He demonstrated cognitive screening tools in dementia and selective supplementary testing from the Behavioural Neurology Assessment that was developed at Baycrest and that is currently being prepared for publication.
Dr. Freedman showed how clock drawing is an easy to administer and sensitive measure of cognitive function. However, not all time settings are equally good for demonstrating deficits. A preferred time to ask the patient to draw is "10 after 11." He gave striking examples in which the clock-drawing test was more sensitive than the Folstein Mini-Mental Status Exam (MMSE). He pointed out that, in conjunction with the clock drawing, mental status testing in the office setting can be effective in defining and tracking the patient's cognitive function.
There are a variety of common clinical problems that are associated with making a diagnosis of dementia.
The first problem is how best to test the patient's memory. Memory is almost always impaired in Alzheimer disease, although poor memory does not necessarily mean AD; intact memory suggests a diagnosis other than AD.
Generally, a patient has a history of memory loss and he or she, and/or the family, is concerned about the possibility of dementia.
The second problem is how best to test the patient's attention. The MMSE uses serial 7s and spelling the word 'world' backwards as tests; however, using months backwards or, in severe cases, having the patient use serial 1s subtraction from one hundred is useful.
A third problem is how to assess language. Dr. Freedman stated that listening to the pattern of spontaneous speech (fluent vs. nonfluent), and testing comprehension, naming and repetition are very useful. Fluent speech suggests a temporal-parietal lesion, and non-fluent points to a frontal lesion. AD produces fluent speech until the later stages.
Auditory comprehension testing involves the assessment of single words, phrases and whole body commands. He suggested that the clinicians also test repetition of single words and phrases, and test naming by showing the patient common objects to name (e.g. pencil and watch). In Alzheimer disease, naming is impaired and repetition is normal in the early stages.
Dr. Freedman outlined testing for ideomotor apraxia, which is the inability to pretend to carry out a motor activity to command (e.g. comb your hair) when the activity is one that can be performed easily in spontaneous situations. To determine if a patient has apraxia, give a verbal command. If the patient fails to respond correctly to the command ask him or her to imitate the action. Finally, ask the patient to use the real object if imitation is impaired.
Visuospatial function is commonly impaired early in the course of AD. Clock drawing is a sensitive test of visuospatial function. Supplementary testing includes drawing to command and copy, such as a house, a flower and a cube.
Dr. Freedman summarized by stating that there are a number of tests of memory. These include asking patients the year, month, day, place, name of the Prime Minister and Premier and immediate and 5 minute 3 word recall words such as cat, apple, table. Attention can be tested through serial 7s and 3s subtraction and by reciting the months backwards. Naming can be tested by asking patients to name objects. Asking the patient to draw a clock, with the time set to 10 after 11, is an effective method to test visuospatial function. Testing similarities and proverb interpretation assesses the ability to manipulate acquired knowledge. It should be noted that the tests of similarities and proverbs may be somewhat confounded by cultural biases. Copying patterns of multiple loops or alternating sequences can be used to assess frontal lobe function. Perseveration on these tasks, and deficits on world list generation of animal names or words beginning with the letter F, are often seen following frontal brain damage.
This workshop reviewed the basic aspects of the mental status exam that make up screening assessment in an office setting. Dr. Freedman concluded that testing cognitive function is very important for the differential diagnosis of dementia.
Dr. Larry Leach presented the next workshop on Neuropsychological Assessment in Dementia. The learning objectives of this lecture were: to determine whether an individual met the basic criteria for dementia; to evaluate the effectiveness of cognitive testing in identifying dementia; and to establish a battery of tests that describes the cognitive profile of a patient with dementia.
The DSM-IV modified definition of dementia was discussed and compared to the definitions provided by Cummings and Benson (1983) and by Strub and Black (1981).1,2
The test battery domains were discussed in terms of memory, abstract reasoning, perceptual functioning, constructional ability, language, praxis, mood, global intelligence and cognitive functioning.
Common referral questions include: a) "Is impairment present?" b)"What is the pattern of impairment?" c)"What is the etiology?" d) "Is it mood related?" d) "And has there been a change?"
Dr. Leach also discussed diagnostic issues involving the effects of age, education, gender and culture.
The Prevalence of dementia was discussed and found to have an incidence of 2.4% in the population aged 5 to 74, 11.1 % in the population aged 75 to 84, and 34.5% in the population over age 85.
Dr. Leach reached several conclusions regarding the use of the MMSE as a diagnostic tool, including that:
a) It was poor for diagnosing dementia when prevalence is less than .60%;
b) It was adequate for ruling out moderate to severe cognitive impairment when prevalence is below .35%, except for those patients who are over the age of 80 and had a lower educational status. Cut-off scores need to be adjusted according to age and education.
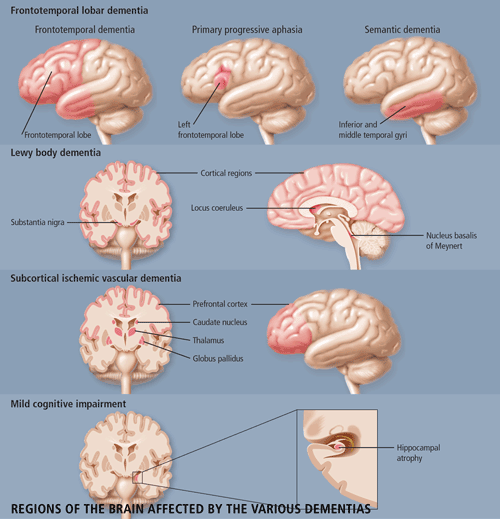
The following criteria for diagnosing 1) Frontal-Temporal Dementia, 2) Lewy-body disease, 3) Vascular Dementia, and 4) Alzheimer Dementia, were discussed. Special tests for dementia were discussed.
Cognitive and neuropsychological tests provide insights into the nature and severity of brain dysfunction as well as brain regions that are dysfunctional in dementia. The pattern of impairment reflects brain regions affected more so than the cause of dysfunction. Therefore, there are practical limitations to diagnosing cause based solely or primarily on the results of mental status examination or neuropsychological assessment. Despite this limitation, the pattern of deficits due to dementia are clearly distinguishable for those cognitive disruptions associated with depression.
Dr. Robert van Reekum followed Dr. Leach with a presentation on the importance of neuropsychiatric evaluation in dementia. He emphasized that changes in mood and behaviour are common in these patients and can cause suffering, impact on disability and handicap, influence diagnosis and have prognostic implications. Perhaps most importantly, these changes in mood and behaviour are treatable. He divided factors that should be assessed into premorbid factors, which included a past history of medical, psychiatric, personal, neurodevelopmental and social factors, and responses to previous treatments, and current factors. The current factors include medical status, arousal antecedents/precipitants/patterns, and cognitive and neurologic status.
A variety of different behaviours are common in patients with dementia. Because the actual dementia may mimic or mask psychiatric disorders, the evaluation of psychiatric illness in this population must take into account the direct effects of CNS disease. Some of the behavioural symptoms of dementia, such as psychosis, may warrant pharmacological intervention. Anxiety, agression, disinhibition and apathy may also warrant treatment.
Finally, Dr. van Reekum stressed the need for structure, reliable and valid Behavioural inventories to improve the consistency of behaviour quantification in these patients. He reviewed one such inventory, the Neuropsychiatric Inventory (NPI).
Dr. Sandra Black presented the pros and cons of the currently available techniques for neuroimaging in dementia. The objectives of this workshop were:
a) To review currently available structural and functional imaging techniques;
b) To review principles for the interpretation of brain-behavior relationships in dementia;
c) To illustrate the above in case examples of patients with dementia.
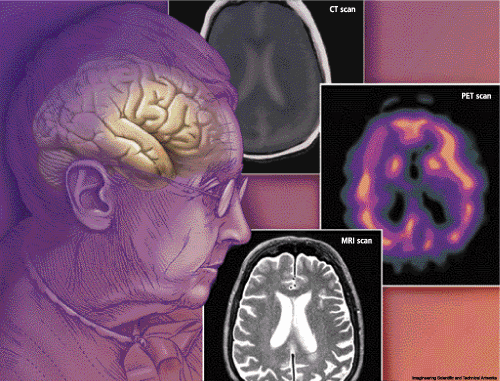
| Currently available techniques for neuroimaging in dementia include magnetic resonance imaging (MRI), positron emission tomography (PET) and CT scan. The relative merits of these techniques were discussed. |
CT scanning in dementia has the following strengths: it has excellent spatial resolution; it is relatively cheap and widely available, it rules out major pathologies; and, if applied correctly, medial temp width can be measured. However, it also presents the following weaknesses: there is less contrast; there is a problem of bone artifact; it is less sensitive to pathology; and the patient is exposed to radiation.
Magnetic resonance imaging (MRI) gives excellent spatial resolution, no bone artifact and better contrast with high sensitivity, and is low risk. However, it has a higher cost than does CT scanning and is less readily available and takes longer to image (but this is changing), and there are associated contraindications (pacemaker, aneurysm and claustrophobia).
Positron Emission Tomography (PET) has the strength of being versatile: injected radiolabel can measure regional cerebral blood flow, metabolism or receptors; direct quantification is possible; there is fair resolution; and it can measure brain activation using subtraction. On the downside, it has a high costs associated with it (cyclotron and special team); it is a scarce resource and not widely available; and there is a risk associated with exposure to radiation.
Single Photon Emission Computed Tomography is relatively cheap and widely available, and the injected radio label measures regional brain perfusion; but it only offers relative quantification, gives poor spatial resolution and again has a risk of radiation exposure.
All of these techniques are useful in different ways for the diagnosis of dementia. Examination of blood flow, oxygen utilization, cerebral atrophy and brain function can be demonstrated using these techniques.
The final lecturer was Dr. Wendy Meschino who discussed the use of Genetic Testing for Dementia in Clinical practice. The objectives of the presentation were how to approach a family history of dementia, risk assessment of hereditary dementia, what tests are available, determining when testing is helpful and providing information on how to get testing done. Alzheimer disease risk factors were identified as: increasing age, a positive family history of AD, Down Syndrome, cognitive impairment, head injury, low education level, and aluminum exposure (controversial). Exposure to exogenous estrogen for women and the presence of arthritis may be protective.
The family history of dementia work-up includes:
a) Taking a detailed three-generation pedigree, noting specific symptoms, such as the age of onset and the number of unaffected relatives;
b) Obtaining medical records, including autopsy, to determine whether the patient suffers from AD or some other condition.
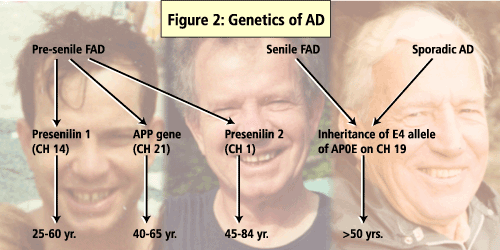
Less than 5% of AD is inherited as an autosomal dominant trait. These cases are usually early in onset. Hereditary factors combined with environmental factors (complex inheritance) play a role in a further 15-25% of mostly late-onset cases. The remaining 75% are sporadic, late-onset and indistinguishable in phenotype from hereditary forms.
Dr. Meschino reviewed a list of genes that are now known to cause hereditary dementias. For early-onset AD these include Presenilin 1, APP and Presenilin 2. Presenilin 1, a gene on chromosome 14, accounts for the majority of early-onset cases. The average age of onset is in the 40's. Some cases of frontotemporal dementia (FTD) are associated with mutations in the tau gene. Notch3 mutations have been found in patients with CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy).
There are also a number of genes that are believed to predispose a patient to late-onset AD. These risk modifier genes are: the e4 allele of the APOE gene on Chromosome 19 (e4 has the effect of decreasing the age of onset), and A2M-2 on Chromosome 12 (in some studies associated with an increased risk of developing AD). These are examples of genes which affect susceptibility to disease, but do not directly cause it. Genetic testing for APOE is not recommended for asymptomatic individuals because the test cannot determine whether an individual will or will not develop AD in the future.
Prior to genetic testing, patients should be provided with genetic counselling, especially when undergoing pre-symptomatic testing. Important information to cover in the session includes outlining the differences between hereditary and sporadic AD, late-onset and early-onset AD, and the risk of developing AD in the general population compared to the risk for that individual.
There are a number of important ethical issues that need to be considered when discussing predictive testing. The patient must be able to make an informed choice; that is, there should be no patient coercion. The clinician should outline the various reasons for knowing or not knowing the diagnosis, the effects it may have on the family and the potential that they will be subject to discrimination. In general, requests for prenatal diagnosis for adult-onset diseases are infrequent, and testing in childhood is strongly discouraged.
One of the following criteria should be met before Alzheimer testing is considered:
a) An individual affected with AD, with onset at less than 60 years;
b) A first-degree, unaffected relative of an affected individual, in a family with 2 or more early-onset cases (all affected are deceased);
c) 2 or more living affected family members with onset greater than 60 years (DNA samples needed from both relatives).
As part of this presentation a video clip was shown from a recent CBC Nature of Things episode called Amanda's Choice, in which a young woman from northern Ontario underwent genetic testing for early-onset Alzheimer disease. There was an extensive family history of the disease in her mother's family with onset of the disease in the mid-30's. She was shown receiving her genetic test results from the presenter as well as genetic counselling. The film explored the emotional impact of living at risk for this devastating disease and the effects on her family.
In summary, these workshops were highly educational and practical. From the neuropsychiatric assessments, MRI and PET diagnostic tests for different dementias, to the ethics and practicality of genetic testing, these workshops appealed to the novice and expert alike.
Dementia: Biological and Clinical Advances--Part II
Dementia: Biological and Clinical Advances--Part III
References
- Cummings JL, Benson DF. Dementia: A Clinical Approach 1983. Butterworths & Company, Canada.
- Strub RL, Black, FW. The Mental Status Examination in Neurology 1981. Philadelphia: FA Davis.