| Christine Oyugi, BSc
Managing Editor,
Geriatrics & Aging.
Edited by:
Karl Farcnik, BSc, MD, FRCPC
Psychiatrist, Division of Geriatric
Psychiatry, University Toronto,
Part-time staff,
Toronto Western Hospital, Toronto, ON. | Contributions from:
Serge Gauthier, MD, FRCPC
Director, McGill Centre for Studies in Aging,
Professor of Medicine (Neurology),
McGill University, Montreal, QC.
Dr. Katarina Rogaeva
Research Associate, Centre for
Research in Neurodegenerative diseases,
University of Toronto, Toronto, ON. |
Introduction
With Canada's aging population, dementia has become a growing problem. Eight percent of Canadians who are over the age of 65 suffer from dementia, of these approximately 60% are believed to have Alzheimer disease. Dementia is an age-related disease, with the prevalence increasing from 2.4% of those from 65-74 years of age, to 34.5% of those 85 and older. There are sixty thousand new cases of dementia diagnosed each year, and the costs of providing health care for these patients continue to escalate. It is with these alarming statistics in mind that clinicians gathered to hear the latest developments in the biology of dementias presented during the 11th annual Rotman conference. Various dementias were discussed at the conference including Frontotemporal lobar dementia (FTLD). FTLD is the third most common form of cortical dementia following Alzheimer disease (AD) and Dementia with Lewy Bodies. It is often mistaken for AD, yet it presents strikingly different clinical and histopathological features and therefore, must be managed distinctly. This article will serve as a summary of some of the key points presented during the first day of this conference on Monday, March 19th.
In her opening remarks, Dr. Rogaeva, a Research Associate at the Centre for Research in Neurodegenerative diseases at the University of Toronto, gave a brief overview of the genetic variability and pathobiology of Alzheimer Disease. AD is a neurological disorder that is characterized by a slow degenerative process affecting cognitive function. At a histopathological level, AD patients are characterized by the deposition of senile plaques within the brain, as well as within the walls of cerebral blood vessels. It is believed that through some unknown mechanism, these senile plaques exert a toxic effect on surrounding neurons, resulting in the neuronal degeneration found in AD patients. Other pathology that is seen includes reactive microglia, swollen neurons and intracellular neurofibrillary tangles.
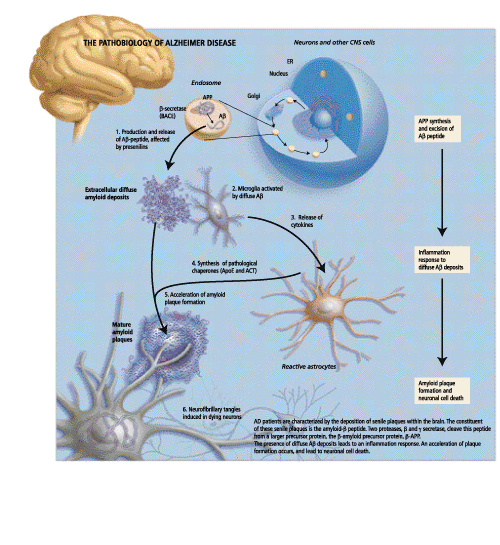
The primary constituent of these senile plaques is the amyloid b peptide (Ab). Two proteases, b-secretase and g-secretase cleave this peptide from a larger precursor protein, b-amyloid precursor protein (b-APP). Essentially, b-secretase cleaves APP to produce an APPsb soluble fragment.
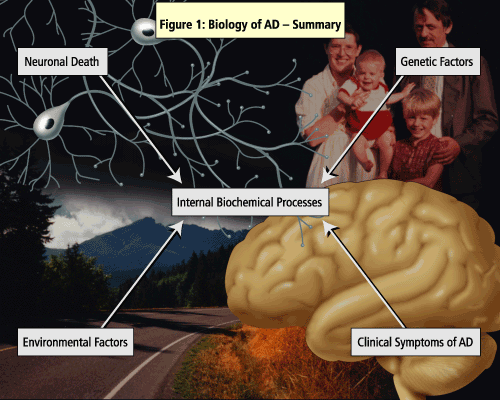
The pathogenesis of AD has been linked to both genetic and environmental factors (See Figure 1).
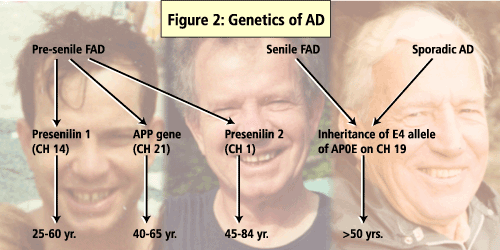
A number of genes have been identified as influencing the development of AD including APO E, the presenilin genes and b-APP gene. (See Figure 2).
Of these, presenilin 1 (PS1) mutations are said to account for majority of the early-onset familial AD cases. In a study that screened AD patients for PS1 mutations, 11% of the cases had mutations in regions coded by PS1 and, of these, 21 were novel mutations. The study also found that the AD patients who had a positive PS1 test were significantly younger (46 ±11yrs) than were the patients who tested negative for PS1 (60±11yrs).
According to Dr. Rogaeva, screening for presenilin mutations is likely to be successful and cost-efficient if it is targeted to the right groups.
The deposition of Ab in the brains of AD patients is linked to the pathology that is characteristic of the disease. In his talk, Dr. Younkin, Director of Research and Professor of Pharmacology at the Mayo clinic in Jacksonville, explained the role of Ab aggregation in the pathogenesis of AD with a focus on whether it is an essential early event in the disease process. One clue to the role of Ab can be garnered from patients with Down Syndrome or trisomy of chromosome 21. These patients invariably develop AD pathology by age 40 and have been found to have high Ab levels in their plasma. Interestingly, this increase in plasma levels of Ab is seen prior to the onset of symptoms. Studies have shown that all mutations that are linked to AD increase the extracellular concentrations of Ab; this phenomenon occurs prior to the development of the disease and fosters Ab aggregation. Aggregated Ab has been shown to be directly toxic to neurons in culture; for this reason inhibition of b or g-secretase, thereby reducing Ab concentrations, has emerged as a therapeutic target for AD. Clinical trials are currently underway to assess the efficacy of immunization with b-secretase.
Plasma and cerebrospinal fluid (CSF) levels of Ab have been shown to increase with age (over age 65 years). A study comparing patients with typical late-onset AD to age-matched controls found that Ab was increased in AD patients. Some of these patients had levels similar to those found in patients with trisomy 21 or familial AD. What is interesting with late onset AD is that, as the disease progresses, there is a decline in the levels of Ab in the plasma and CSF. The reason for this decline is not well understood, although microglial clearance has been implicated.
Although plasma levels of Ab are not adequate for making a diagnosis of AD, they may be useful as a biomarker for the disease. According to Dr Younkin, in the future, therapies could be targeted to patients with elevated Ab in the same way that patients are treated for elevated cholesterol levels to prevent cardiovascular disease. The study of Ab is only one of many approaches to the study of AD. Other approaches could play important roles in the disease progression and could also be key targets for therapeutic intervention.
Dr. Wilhelmsen, Associate Professor of Neurology at the University of Carlifornia, gave a brief overview on the pathobiology of taupathies. His talk was based on clinical evidence gained from a family with members that suffered from a variety of neurodegenerative diseases. Some of the symptoms observed included: behavioural disinhibition, dementia--which differed from that seen in AD in that there was a relative preservation of language and praxis--and Parkinson's disease, without the typical tremor and non-responsive to L-DOPA. Most members of the family with the disease eventually developed amyotrophy--which is the loss of motor neurons resulting in the development of brisk reflexes, but reduced motor power in the limbs. However, by the time that most patients were dying they were all akinetic, in a fetal position and bed-ridden, making it difficult to recognize that they had typical signs of motor neuron disease. Personality changes were observed in family members including aggressiveness, depression, alcoholism, hyper- and hypo-sexuality, childishness, craving for sweets and hyper-religiosity. Interestingly, they did not respond very well to neuroleptics. The age of onset of symptoms ranged from 25-56 years.
At the time of autopsy, microvascular changes were observed in the anterior temporal cortex of the frontal lobes, between layers II and III. Other pathology includes, rare ballooned neurons, swollen vacuolated anterior horn cells, loss of pigmented cells in the substantia nigra and gliosis in the hippocampus.
Linkage analysis was performed and a link was found to chromosome 17. This analysis was done on several families all of whom had suffered from a variety of disorders including progressive Parkinsonism and dementia with palido-ponto-nigral degeneration (PPD), frontotemporal dementia and primary progressive aphasia (PPA). Several of these families had mutations in the tau gene, which has been implicated in the formation of neurofibrillary tangles in AD. Tau protein deposits have been linked to a variety of neurodegenerative diseases, many of which are frontotemporal dementias or movement disorders, collectively referred to as tauopathies--Pick's disease, progressive supranuclear palsy, and corticobasal degeneration.
What is the function of tau? The tau gene is large and has a complex pattern of alternative spicing. The protein has domains that have been shown to affect the assembly of microtubules. Disruption of this alternative splicing is enough to cause a dysfunction of the tau protein and resulting neurodegeneration. It appears that the regulation of splicing of the gene is important for maintenance of normal brain function. Future research is needed to elucidate whether it is the aggregation of the protein that ultimately results in disease.
Many doctors have never seen a case of progressive supranuclear palsy (PSP). The disease is often misdiagnosed as Parkinson's disease until the patient fails to respond to normal therapy for PD. The prevalence of the disease is less than that for PD. The pathology of PSP is marked by the precipitation of tau protein throughout the midbrain. Although PSP is primarily a sporadic disease, studies show that a precipitating mutation may be required for the disease to occur.
Aggregates of tau protein are seen in many neurodegenerative disorders suggesting that this process may be a common pathway in the pathology of these diseases. Although the importance of the tau gene is known, more research is needed into the interaction of tau with other genes, as well as the regulation and metabolism of tau. Future efforts to develop animal models of tau-mediated neurodegeneration should provide further insights into the onset and progression of tauopathies, as well as Alzheimer disease. This could lead to the discovery of effective therapies for these disorders
The next speaker was Dr. David Westaway who began his review of the latest research in prion diseases. Prion diseases, or transmissible spongiform encephalopathies, are fatal neurodegenerative disorders. These neurodegenerative diseases include scrapie in sheep, mad cow disease in cattle, and Creutzfeldt-Jakob disease (CJD) in humans. Prion diseases may present as genetic, infectious or sporadic disorders, all of which involve the post-translational modification of the prion protein (PrPC) into the disease-causing PrPSc. CJD generally presents as progressive dementia where as the other forms of prion disease typically manifest as ataxia-like disease.
More than 20 mutations of the PrPC gene are now known to cause the inherited human prion diseases, and significant genetic linkage has been established for five of these mutations. However, why or how these mutations cause the protein to change and result in disease is not really understood.
The function of PrPC is still unknown, although there is some evidence that it is involved in the binding of Cu(II) ions. Recent studies suggest that PrPC may function in signal transduction through a pathway involving Fyn tyrosine kinase. Fyn is associated with acetylcholine receptors in muscle cells. It is highly expressed in the limbic system and may have a role in myelination. It was hoped that deletion of the mouse Prnp gene would alter the phenotype such that conclusions could be made with respect to the function of PrPC. Studies in mice show that injection with PrPSc results in death within 150 days. However, Prnp0/0 knockout mice are resistant to prion infection. This provides evidence that cellular PrP is necessary for disease pathogenesis.
Dr. Sandra Black then gave an overview of the use of neuroimaging biomarkers for AD. An ideal test for AD should: reflect the pathophysiology of the disease, allow for pathologic evaluation, allow for early detection, differentiate AD from other dementias, be non-invasive, be affordable, have a sensitivity greater than 80% for detecting AD and a specificity of greater than 80% for distinguishing other dementias. The desired sensitivity and specificity of a biomarker depends on its purpose.
Dr. Black suggested that current clinical techniques can be termed the "bronze standard" for diagnosis. This is based on the combined use of history, and physical and cognitive examination, as well as blood test and neuroimaging to exclude secondary causes. The "gold standard" is based on tissue pathology. Biochemical markers for AD include apolipoprotein E (ApoE) genotyping, and several potential CSF markers including: beta-amyloid, possibly reflecting amyloid deposition and formation of senile plaques; PHFtau protein as a marker for the phosphorylation state of tau, and formation of neurofibrillary tangles; (total) tau protein, a normal axonal protein, used as a marker for ongoing neuronal and axonal degeneration, and the CSF/serum albumin ratio, as a marker for blood-brain barrier damage, used to exclude patients who also have cerebrovascular pathology.
Functional imaging techniques such as PET and SPECT also serve as important diagnostic tools. Recently, functional studies have shown abnormalities in the posterior cingulate and medial temporal regions of patients who show memory impairment and later develop AD. This finding is potentially useful in detecting pre-clinical AD, but it is difficult to apply to individual cases.
With the emerging therapeutic compounds for the treatment of AD, an important role of imaging is in monitoring whether treatment is actually slowing down the progression of atrophy. Several techniques have been proposed to monitor the progression of the atrophy, but currently, none can be performed reliably. It is likely that both quantitative neuroimaging and biochemical profiles (urine and serum) together with clinical neurobehavioural assessments will be needed in order to achieve the required sensitivity for diagnosing and monitoring AD. Dr. Black stressed the urgent need for sensitive and specific tests.
Dr.Serge Gauthier gave the final talk of the day on currently available and future therapies for AD and related conditions. In his opening remarks, Dr. Gauthier stressed that it is important for clinicians to be knowledgeable of the fact that AD is not just one condition, but entails a mild stage AD, moderate AD and severe AD. Different treatments are effective at different stages in the disease.
In typical cases, AD progresses through relatively predictable stages, as described in the Global Deterioration Scale of Reisberg et al., (Reisberg B, Ferris SH, Deleon MJ, Crook T. The global deterioration scale for assessment of primary degenerative dementia. Am J Psychiatry 1994;44:2203-6). The estimated time from diagnosis to death is usually 5-8 years. In typical AD, most patients initially present with a change of mood, which improves over time. They then experience a linear cognitive and functional decline including a loss of autonomy for instrumental and self-care activities of daily living. Most patients have some degree of neuropsychiatric change, including hallucinations, misidentifications (Capgras syndrome), delusions and paranoid ideation, aggression or apathy, wandering and sexual disinhibition. The appearance of early-onset motor or gait disturbances, including asymmetrical grasp responses is atypical and suggests conditions other than AD. Dr. Gauthier described milestone steps in the progression of AD. These are important as they may act as future therapeutic targets (Table 1).
| TABLE 1 Milestones in Progression of AD |
| a) Diagnosable dementia
b) Loss of IADL (Instrumental Activities of Daily Living)
c) Emergence of neuropsychiatric symptoms
d) Nursing home placement
e) Loss of basic ADL
f) Death |
Different stages of AD present different issues in management of the disease. In the early stages of AD the management issues include making an accurate diagnosis, patient and caregiver education, advance power of attorney, advance directives and whether the patient should continue driving. In the moderate stages, the physician should carefully monitor the patient's autonomy. At this stage it is very important to monitor the health and well-being of the caregiver. Management issues in the final, severe stages of AD include cessation of cholinesterase inhibitors and end-of-life decision making.
Taken together the speakers presented a great deal of useful information on the pathobiology of a variety of neurodegenerative diseases. Understanding the genetic and biochemical basis of these diseases may allow for treatments tailored to various disease stages, as well as providing useful biomarkers to enable detection of those patients who are most at risk.
Dementia: Biological and Clinical Advances--Part I
Dementia: Biological and Clinical Advances--Part III